Phylogeny Exercise¶
Author: Vi Varga
Course: Applied Bioinformatics (BBT045)
Due Date: 03.03.2025
Grading¶
This assignment is graded on a pass/fail basis. In order to pass, you must obtain at least 60% of the possible points (i.e., a minimum of 9 points out of the maximum possible 15 points).
Proper code documentation is worth 0.5 points, and the inclusion of all supplementary files is worth an additional 0.5 points. Otherwise, the point values are written next to each question. You will notice that questions that require data interpretation are worth more than those that require straightforward/descriptive answers.
Introduction¶
In this exercise you will practice your newly acquired skills in phylogenetic inference and "tree thinking", by analyzing the evolutionary history of a gene family. Please note that unlike the tutorial, this exercise should be completed on your own. If you have questions, please reach out to me via e-mail (virag.varga@chalmers.se) or message me on Canvas. The assignment should be uploaded to Canvas as a Jupyter Notebook (.ipynb file) by 23.59 on Monday, March 3rd, 2025, along with the requested supplementary files.
If you do not already have this Jupyter Notebook downloaded, you can use wget to download the Jupyter Notebook version of this file from here. If you wish to be able to see the images referenced in this Notebook, please copy the contents of the /cephyr/NOBACKUP/groups/bbt045_2025/Resources/Phylogeny/Exercise/ directory to a directory in your local/home directory, ideally at PhyloWorkflow/Exercise/. You should use the same runtime environment, phylogeny-jupyter.sh, for this exercise, as you used for the associated tutorial.
In this exercise you are going to follow the phylogenetic analysis workflow discussed during the lecture and tutorial, starting with collecting the data necessary for running the analysis (in the form of homologous protein sequences from different species), through the interpretation of the results (i.e., comparing species trees and gene trees).
For this exercise, you will be working with species within a phylum of protists (single-celled eukaryotes) known as the Apicomplexans. Apicomplexans are mostly obligate parasites, meaning that they rely entirely on their host organism to survive. There is debate over the possibility of some species from this phylum being host-associated but non-parasitic, and some recently-discovered Apicomplexans are thought to be free-living, though they are not well-studied (del Campo et al. 2019).
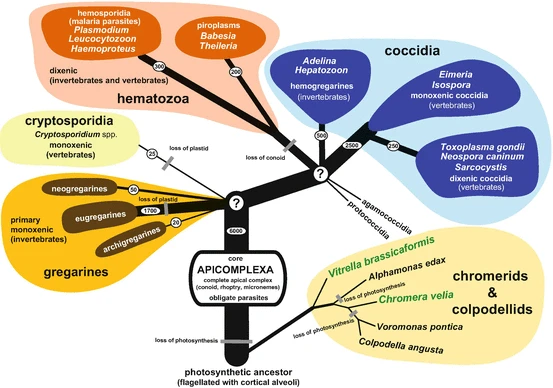
Source: Votýpka et al. 2016
A Newick tree for the clades of interest is provided below (as well as in the Apicomplexa_clades.nwk file):
(Gregarinasia,(Cryptosporidia,((Piroplasmida,Haemosporidia),Coccidia)));
When visualized as a cladogram, the tree looks like this:

Below are listed the species you will be working with:
- Porospora gigantea
- Plasmodium falciparum
- Babesia microti
- Toxoplasma gondii
- Cryptospodidium parvum
- Which clade of Apicomplexa do each of the above species belong to? List them below. (0.5 points)
Fill in your answer!
You might recognize some of these names! Phylum Apicomplexa contains a number of medically important parasites:
- Cryptosporidium species cause Cryptosporidiosis, a water-borne diarrheal disease that often affects immunocompromised people.
- Babesia species cause Babesiosis, a tick-borne illness that causes flu-like symptoms. Since the parasite infects and destroys red blood cells, it can also cause anemia.
- Toxoplasma gondii causes the disease Toxoplasmosis, which humans contract from food and/or being around pets that are infected. Most infections are asymptomatic, though it can cause flu-like symptoms and serious complications for the fetus if contracted during pregnancy, up to stillbirth or miscarriage.
- Plasmodium falciparum is a causative agent of Malaria, a disease which still kills more than half a million people each year. Among the Plasmodium species, P. falciparum causes the most deadly strain of the disease.
The Gregarines are comparatively mild parasites - they infect invertebrates, and are rarely fatal to the organisms they infect.
- Why might it be important to study organisms like the Gregarines? (Hint: Take another look at the phylogenetic tree of Apicomplexa.) (0.5 points)
Fill in your answer!
Before moving on to the actual phylogenetic workflow, you should also decide what the outgroup will be. This won't actually come into play until the visualization stage, but it's always good practice to be thinking ahead when following a research pipeline.
- Which group of organisms should be the outgroup? Why? (0.5 points)
Fill in your answer!
You may be wondering, "Why not select an outgroup from elsewhere in Alveolata (the superphylum that Apicompla belongs to)?" After all, the Gregarines are considered part of Apicomplexa - aren't they too closely related to the rest to be useful in a study like this?
In most cases, you might be right! A group of organisms like those in Chromerida, which are also in Alveolata but not considered Apicomplexans, might be a better choice!
(Chromerida,(Gregarinasia,(Cryptosporidia,((Piroplasmida,Haemosporidia),Coccidia))));

In this case, we'll be using the Gregarines because of the gene families presented to you as options - all of these gene families are involved in the "glideosome," which is a key component of Apicomplexan pathogenicity. The glidesome is involved in parasite attachment to, movement along and invasion of host cells. More distantly related species within Alveolata may not have copies of these genes. For example, C. velia doesn't have a copy of the ROM4 gene (Boisard et al. 2022).
Obtaining & Exploring Data¶
For this exercise, you will be obtaining your own data from the NCBI database.
You will start by selecting a gene family to study. Below I've listed a few options:
- ROM4
- AKMT
- MyosinA
- DGK1
Since Plasmodium falciparum and Toxoplasma gondii have really well-annoted genomes, search for the gene sequence of your choice on the NCBI with, for example: "Plasmodium falciparum[orgn] ROM4". Save the header and amino acid sequence to a FASTA file on your local machine. I've provided recommended search combinations below to make things go a bit more smoothly; the members of these protein families do not all include these gene family names in the actual names of the individual genes themselves on the NCBI. If there are multiple results, just go with the first gene hit.
Search recommendations:
- ROM4: P. falciparum ROM4
- MyosinA: P. falciparum Myosin-A
- AKMT: T. gondii apical complex lysine methyltransferase
- DGK1: T. gondii diacylglycerol kinase accessory domain
Save the FASTA sequence of your selected reference protein representative of your chosen protein family to a file on your local computer. This file should be included in your assignment submission.
Next, you'll need to obtain homologous gene sequences from the NCBI database. BLAST your protein sequence on the web to accomplish this, and choose the "FASTA (complete sequence)" download option. This file should be included in your assignment submission.
Note that you can select specific organisms to BLAST against on the NCBI - this significantly reduces the amount of time a search takes, and in our case, makes sure you get only the information you're looking for. There are of course many members of the Apicomplexa that we will not be working with here - no need to sift through all of the gene sequences from those organisms. Include only our 5 species of interest; note that you'll need to select both the A and B versions of P. gigantea individually.
- Open your multiple sequence FASTA and describe some of its characteristics. How many sequences are in the file? How many representatives of each species are there (count the two P. gigantea species as only one)? (0.5 points)
Fill in your answer!
- Describe the following examples with all words that apply from among: homologous, orthologous, paralogous (2 points)
a. Two copies of MyosinA in B. microti
Fill in your answer!
b. Copies of AKMT in P. falcigarum and T. gondii
Fill in your answer!
Before proceeding on to the construction of the MSA, clean your sequences.
- Do you notice any differences in the files? What does this tell you? (0.5 points)
Fill in your answer!
Multiple Sequence Alignment¶
Now that you've familiarized yourself with the data, it's time to create a multiple sequence alignment.
- Write the code you used to create the MSA below. Explain the meanings of the flags, and why you chose to use them. (0.5 points)
Fill in your answer!
#Fill in your code here!
This MSA file should be included in your assignment submission.
Now open the MSA file in your alignment viewer of choice.
- What patterns do you observe? (1 point)
Fill in your answer!
Creating the Tree¶
Now it's time to create a phylogenetic tree. Since you're less pressed for time than you were in-class, feel free to use the model finder option of IQ-TREE, if you wish to try it out. However, if you'd prefer to use the same tree model as you did during the tutorial, that's fine, too! Write the code below:
#Fill in your code here!
- Which tree model did IQ-TREE pick for you, if you used the model finder? Why is the model finding process important? (2 points)
Fill in your answer!
The PREFIX.treefile and PREFIX.log files should be included in your assignment submission.
Now open the PREFIX.treefile in a phylogenetic tree visualizer of your choice.
- What does it look like to you? Do you notice anything? (0.5 points)
Fill in your answer!
Edit the alignment one time, either manually or using a tool. Save the new MSA, and run IQ-TREE on it again, using the model that IQ-TREE previouslsy determined to be the best fit for you. The new MSA file, PREFIX.treefile and PREFIX.log files should be included in your assignment submission.
- What method of alignment editing did you choose to use? Does your alignment look different in an alignment viewer? Does the resulting tree look different? How so? If you used a command-line tool, include the code below. If you used an interactive editor, describe your strategy. (1.5 points)
Fill in your answer!
#Fill in your code here!
Comparing the gene tree with the species tree¶
Finally, compare the two gene trees you have visualized with the species tree given to you. (The Python script provided to you during the tutorial may help with this, but feel free to do it your own way!) Include .png/.svg/.jpeg/etc. versions of both of your visualized phylogenetic trees in your assignment submission.
- How do the gene trees compare to the species tree? Do you notice any interesting patterns? (2 points)
Fill in your answer!
- Is this a good gene family for creating a species tree? Why or why not? (2 points)
Fill in your answer!
Citations¶
Boisard, J., Duvernois-Berthet, E., Duval, L., Schrével, J., Guillou, L., Labat, A., le Panse, S., Prensier, G., Ponger, L., & Florent, I. (2022). Marine gregarine genomes reveal the breadth of apicomplexan diversity with a partially conserved glideosome machinery. BMC Genomics, 23(1), 1–22. https://doi.org/10.1186/S12864-022-08700-8/FIGURES/5
del Campo, J., Heger, T. J., Rodríguez-Martínez, R., Worden, A. Z., Richards, T. A., Massana, R., & Keeling, P. J. (2019). Assessing the diversity and distribution of apicomplexans in host and free-living environments using high-throughput amplicon data and a phylogenetically informed reference framework. Frontiers in Microbiology, 10(OCT). https://doi.org/10.3389/fmicb.2019.02373
Töpel, M. (2019, November). Webbased Phylogenomic analysis · The-Bioinformatics-Group/Teaching Wiki. GitHub. https://github.com/The-Bioinformatics-Group/Teaching/wiki/Webbased-Phylogenomic-analysis
Votýpka, J., Modrý, D., Oborník, M., Šlapeta, J., & Lukeš, J. (2016). Apicomplexa. In Handbook of the Protists (pp. 1–58). Springer International Publishing. https://doi.org/10.1007/978-3-319-32669-6_20-1